A brief history of optical microscopy
Saturday 16 May is the international day of light.
To celebrate we sat down with Sergio Lopez from our Bioimaging facility for a brief overview of microscopy, from a flask of water to our latest high-tech addition, the Stellaris microscope.
“As you might imagine, light is essential to many forms of bioimaging. For instance, light microscopy uses visible light to create magnified images of minute objects and differs from electron microscopy, which relies not on visible light, but on electron beams to create its magnified images.
The use of light for the magnification of small objects has a long, rich, and interesting history.
The first magnifiers
One of the first attempts at using light for this purpose was that of Seneca, who in the 1st century AD noticed that a flask filled with water could be used to create magnified images of small letters. This discovery eventually led to the creation of reading aids that made use of spherical pieces of transparent minerals.
In the late 13th century, in Italy, clear silicate glass was produced for the very first time. As a result, both concave and convex lenses of relatively high quality started to be manufactured, paving the way for the construction of the first simple microscopes.
A simple microscope is a microscope that consists of a single lens, much like the magnifiers used by Sherlock Holmes.
Compound microscopes
The light microscopes that we use today are not simple microscopes, but compound microscopes, comprised of two or more lenses.
The first compound microscopes were manufactured in the Netherlands at the end of the 16th century. It is not clear who built the first one, but according to the available evidence it seems that it was Zacharias Janssen, a spectacle-maker from Middleburgh.
Compound microscopes should be much better instruments than simple microscopes because, at least in theory, they should be able attain larger magnifications. However, the first compound microscopes were plagued by optical aberrations that seriously compromised the quality of the images.
For this reason, up until the 19th century simple microscopes outperformed compound microscopes.
For example, Robert Hooke’s compound microscope, made in London by Christopher White circa 1670, although good enough to allow Hooke to become the first person in history to observe microorganisms, was surpassed in performance by the simple microscope that was built a few years later by Antoine van Leeuwenhoek, a draper from Delft.
Initially, van Leeuwenhoek’s intention had been to obtain magnified images of the threads he had in his draper shop, but he soon realised that his microscope also allowed him to see minuscule beings that he named animalcules. These animalcules were what we now call unicellular organisms, such as bacteria and protozoa.
When the optical aberrations that impaired the performance of compound microscopes were successfully resolved in the first half of the 19th century, light microscopy experienced a huge leap in image quality and details that had hitherto been invisible could finally be observed with clarity.
The improved compound microscopes were instrumental in the development of the cell theory, which was enunciated in 1839 by Schleiden and Schwann. According to this theory, which was later corroborated innumerable times, both animals and plants are made up of fundamental units of life called cells.
The compound microscope continued to improve in performance during the second half of the 19th century, with many of the innovations were introduced by Ernst Abbe, a professor at the University of Jena and co-owner of Carl Zeiss AG.
In addition to coming up with inventions such as the apochromatic lens system, Abbe was the first to realise that due to the very nature of light, and in particular due to a phenomenon known as diffraction, light microscopes would never be able to distinguish between objects separated by a distance of 200 nanometres or less. In other words, two objects separated by such a distance would always look like a single object under a light microscope.
This distance, which became known as the Abbe diffraction limit, is roughly 10,000 times smaller than the diameter of the head of a pin. It would take another 100 years for a light microscope to break the Abbe diffraction limit.
Fluorescence
In the 20th century, scientists started developing radically new light microscopy techniques, one of which relied on a phenomenon called fluorescence.
When a fluorescent substance is illuminated with light of the appropriate colour, it will emit light of a different colour. For example, quinine sulphate, which is present in tonic water, emits blue light upon being illuminated with ultraviolet light.
Since the light used for illumination and the light emitted by the specimen (i.e., the fluorescence) are of a different colour, they can easily be separated from each other using optical filters.
With the right setup, a microscope can therefore reject the light coming from the illumination source and use only the light emitted by the fluorescent sample to form an image. This results in an image that has a very good contrast and whose intensity can be used for quantitative determinations.
The first fluorescence microscopes were built at the beginning of the 20th century by the German company Carl Zeiss AG and the Austrian company Carl Reichert.
Confocal microscopes
Despite the advantages of fluorescence microscopy, a serious problem remained.
Fluorescence microscopy images were formed not only by light emitted at the plane of interest, but also by light emitted at planes above and below. This out-of-focus light caused fluorescence images to be somewhat blurred.
The solution was to introduce a small orifice called the pinhole into the optical path.
The pinhole, which is placed in a very precise location within the body of the microscope, rejects the fluorescence coming from out-of-focus planes, but allows the fluorescence coming from the focus plane to pass through and reach the detector.
By using a pinhole, the resolution of the microscope is greatly improved, especially in the direction that is parallel to that of the illuminating beam.
A microscope equipped with a pinhole is called a confocal microscope.
The first confocal microscope was built by Marvin Minsky in the mid-1950s.
However, it would take roughly 30 years for confocal microscopy to gain popularity within the scientific community, as only with the invention of the laser and the advent of suitable computers would confocal microscopy reach its full potential.
Breaking the diffraction limit
Starting in the 1990s, several fluorescence microscopy techniques capable of breaking the Abbe diffraction limit were developed, known as super-resolution microscopy techniques.
These can be classified into two groups.
- The first group includes techniques based on using complex illumination patterns, such as donut-shaped laser beams or beams forming periodic patterns. Examples of these techniques are structured illumination microscopy (SIM) and stimulated emission depletion microscopy.
- The second group includes techniques based on single molecule localisation. These techniques involve acquiring a large number of images, each of which contains only a few isolated fluorescent molecules. The location of these molecules is then determined with great precision using mathematic algorithms. Finally, all of the locations are combined into a single image of superb resolution. Examples of single molecule localisation techniques are photoactivated localisation microscopy (PALM) and stochastic optical reconstruction microscopy (STORM)
The John Innes Centre Bioimaging Facility offers its users the possibility of implementing SIM, PALM, and STORM by using the Zeiss Elyra PS1 microscope.
Fluorescence lifetime imaging microscopy
Another state-of-the-art fluorescence microscopy technique is fluorescence lifetime imaging microscopy (FLIM).
Each pixel in a FLIM image contains information not only about the fluorescence intensity emitted by the specimen, but also about the time that it took for the specimen to emit that fluorescence. This time can be influenced by environmental parameters like viscosity, pressure, pH, ion concentrations, and others.
Usually, it takes only a few nanoseconds for a fluorescent molecule to emit light (a nanosecond is a billionth of a second). Therefore, FLIM requires highly specialised detectors and electronics.
Very recently, new and improved microscopy components were developed that transformed FLIM from a slow technique into a relatively fast technique.
At the moment, very few imaging platforms in the world are capable of performing ultrafast FLIM. The John Innes Centre Bioimaging Facility is one of them however, thanks to our most recent purchase of the Leica Stellaris 8 microscope.
This international day of light will be a good day to remember all that has taken place in the field of light microscopy, from Seneca’s water-filled flask to super-resolution microscopy.
It will also be a good time to think about the fact that, thanks to the cutting-edge microscopes of the John Innes Centre Bioimaging Facility, you might be the one to write the next page in the fascinating history of light microscopy…”
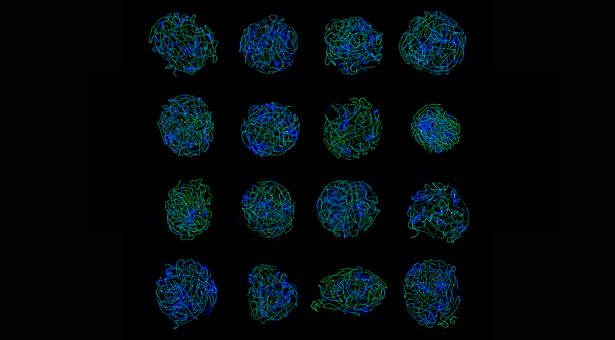
The image at the top of this page shows a collage of structured illumination microscopy (SIM) images acquired with the Elyra PS1 microscope. The cells are Arabidopsis arenosa prophase I cells. The synaptonemal complex (ZYP1) is shown in green, the crossover sites (HEI10) are shown in red, and the DNA (DAPI) is shown in blue. The image was kindly provided by Dr Chris Morgan from Dr Xiaoqi Feng’s group.


