Epigenetic memory systems
Understanding the mechanistic basis of epigenetic memory is a central question in molecular biology.
A key hypothesis is that covalent modifications, such as trimethylation, added to specific locations on histone proteins are one important epigenetic memory element.
Although much is known about the proteins involved in histone modification dynamics, our overall mechanistic understanding is still limited.
Recently, the Martin Howard lab has been working closely with Caroline Dean’s lab in an effort to more fully understand these issues in the context of the floral regulator gene FLC. FLC is repressed by cold, such that in a subsequent warm period, the level of repression is quantitatively dependent on the prior cold period.
In effect the length of the cold period is epigenetically remembered by the plant, a memory that is crucial in the timing of flowering and therefore of reproductive success.
The group have shown that this quantitative epigenetic memory is encoded in an all or nothing fashion at each locus (where FLC is either fully repressed or not), with the fraction of repressed loci increasing with an increasing period of cold (Angel et al, 2011, Berry et al, 2015). In other words the epigenetic response is fundamentally digital:
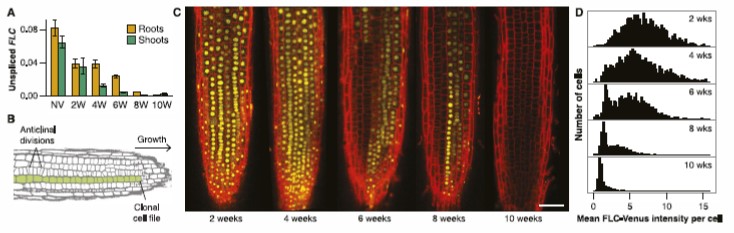
More recently using dual-fluorescent labelling, the Howard group have shown have shown that the epigenetic memory itself is stored in the chromatin and not in networks of diffusible trans factors.
This is a critical result as it demonstrates at the level of a single gene that epigenetic information can be stored locally in, for example, histone modifications. However, the information that can be stored at a single locus is very limited, only 1 bit, ON or OFF.
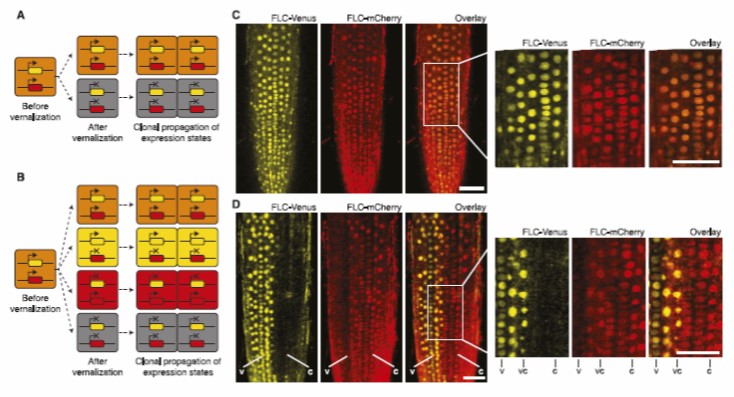
Although based on the FLC system, the high levels of conservation of epigenetic elements, means that the conclusions from our work may be very widely applicable in biology.
Transcriptional regulation
The textbook view of how transcription is quantitatively regulated is through changes in transcription initiation. However, the arrangement of DNA in chromatin in eukaryotes and the frequent occurrence of noncoding transcripts add to the complexity of transcriptional regulation.
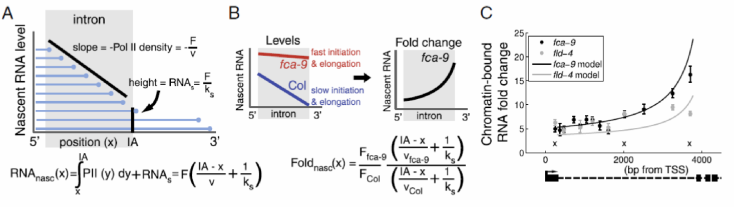
Recently, the Howard group have explored the quantitative non-epigenetic transcriptional regulation of FLC. Prior to cold exposure, FLC expression correlates with altered antisense transcript processing and different chromatin states.
Through experiments and mathematical modelling, the group have discovered that transcription initiation and elongation are tightly coordinated and both are influenced by the chromatin state at the locus. Indeed, modulation of the chromatin environment by noncoding transcripts to coordinately influence transcription initiation and elongation could be a general mechanism to regulate quantitative transcriptional output. Modulation of elongation rates could be particularly important in cases where kinetic coupling can lead to differential selection of splicing isoforms.
Digital versus analogue control
A key question to answer is how digital epigenetic regulation is combined with more conventional analogue transcriptional control, provided, for example, by transcription factors.
This is not straightforward, as digital regulation relies on strong positive feedback, which may conflict with more fine-tuned analogue transcriptional control.
Understanding how these two fundamentally different modes of regulation, digital versus analogue, can be combined is now a key goal.
The Howard group is trying to answer these questions both at Arabidopsis FLC, and also in embryonic stem cells, in a new collaboration with the experimental group of Prof. Rob Klose (Oxford University).